continuidade ao seu tratamento. Sobre esta doença e suas respectivas características, analise.
I. A Síndrome de Guillain-Barré (SGB), doença neurológica capaz de provocar fraqueza muscular generalizada que, em alguns casos, pode levar à morte, caracteriza-se como autoimune, com predominância maior em homens na idade de 20 a 30 anos.
II. Conduz à desmielinização da bainha de mielina e a degeneração axonal dos nervos periféricos.
III. Acarreta paralisia espástica, arreflexia ascendente e diferença na concentração de proteína no líquido cefalorraquidiano (LCR), caracterizando a dissociação albumino-citológica, que é característica da SGB.
IV. Os pacientes com SGB apresentam muita dor, descrita como parestesia, disestesia, dor axial e radicular, mialgia, dor articular e desconforto visceral. A fisioterapia motora constitui-se de exercícios passivos, ativoassistido e ativo dos membros, este último dependendo da melhora da fraqueza muscular.
Estão corretas apenas as afirmativas
A) I e IV.
B) III e IV.
C) I, II e III.
D) I, II e IV.
E) II, III e IV.
Pessoal questão muito interessante.Todas as opções estão corretas, apenas a “III” está errada, ela propõe a paralisia espástica, o que é incorreto.
Coloquei no final desse post o depoimento muito interessante de uma paciente. Ao nos aproximarmos das patologias numa situação real, ao vivenciarmos o que ocorre, podemos fixar melhor o conhecimento.
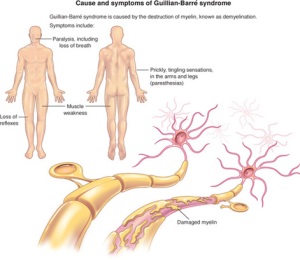
A Síndrome de Guillain Barré (SGB) é uma doença autoimune geralmente desencadeada após uma infecção viral ou bacteriana (ainda que também possa ocorrer após vacinação, cirurgia, anestesia epidural, transplante de órgãos e medula óssea, linfomas, sarcoidose e uso de penicilina), que provoca paralisia ascendente (dos pés até a cabeça) progressiva e potencialmente fatal.
Descrita inicialmente por Jean B. O. Landry em 1859, médico francês, como um distúrbio dos nervos periféricos que paralisava os membros, o pescoço e os músculos respiratórios. Em 1916, os médicos parisienses: Georges Guilliain, Jean Alexander Barre e André Strohl, demonstraram a anormalidade característica do aumento das proteínas com celularidade normal, que ocorria no líquor dos pacientes acometidos pela doença. Eis porque esta síndrome também é chamada de Landry-Guillain-Barré.
EPIDEMIOLOGIA
É uma doença cosmopolita de incidência anual entre 0,4 a 4 casos/100.000 habitantes (média de 1,3).Esta síndrome acomete todas as idades; no entanto, alguns estudos epidemiológicos mostram um pequeno pico na adolescência e adulto jovem, provavelmente devido ao maior risco de infecções.
Algumas séries mostram homens mais susceptíveis que as mulheres em torno de 1,25: 1. Desde que se iniciou a vacinação contra poliomielite a SGB tornou-se a causa mais frequente de paralisia flácida aguda.
CAUSAS
A SGB é de provável etiologia autoimune que pode ser desencadeada após 15 a 21 dias, em média, de uma infecção do trato gastrointestinal ou trato respiratório. Após o processo infeccioso o organismo produz anticorpos que agridem as bainhas de mielina de nervos perífericos e raízes nervosas. A mielina é a estrutura responsável por proteger (como uma capa) e permitir a transmissão do sinal nervoso ao longo de todo o trajeto do nervo.
Isto provoca fraqueza (paralisia flácida) e insensibilidade, que caracteristicamente progride de baixo para cima, em direção à cabeça, ascendente. O agente mais comum encontrado como causador é o Campylobacter jejuni, que provoca infecção gastrointestinal. Há também relatos de casos com Citomegalovirus, Epstein Barr vírus, Micoplasma pneumoneae.
Esta síndrome também é reconhecida como uma das complicações pela infecção pelo HIV, e também há relato de casos em decorrência de vacinação, cirurgia, anestesia epidural, transplante de órgãos e medula óssea, linfomas, sarcoidose e penicilina. Parece que a gravidez e uso de anticoncepcional oral conferem algum grau de proteção.
QUADRO CLÍNICO
Primeiramente é preciso esclarecer que a SGB não tratada leva rapidamente à morte, portanto, a constitui uma emergência médica, inclusive com indicação de tratamento em Unidade de Terapia Intensiva – UTI.
Doença de instalação hiperaguda – horas ou dias.
Sintomas mais frequentes: formigamentos e agulhadas nos pés; dores surdas nas costas; fraqueza muscular.
Inicia-se com dores lombares associadas a desestesias nas extremidades inferiores.
Com a progressão os membros inferiores ficam paréticos ou plégicos.
Ocorre paraparesia motora e sensitiva (paralisia incompleta), paraplegia flácida, e arreflexia.
Os reflexos tendinosos são perdidos precocemente, mesmo nas regiões onde a força muscular está preservada.
O quadro progride de modo ascendente: dos pés para cabeça.
50% dos casos pode acometer a face e função bulbar: disparesia parcial periférica, disfagia, disartria.
30% dos casos: paralisia da musculatura respiratória.
Exame Físico:
Paresia ou plegia
Arreflexia
flacidez simétrica
Sensibilidade térmica e dolorosa preservadas.
Sensibilidade vibratória e proprioceptiva comprometidas.
Em pacientes graves: insuficiência ventilatória, disautonomias (arritmias cardíacas, sudorese, picos hipertensivos).
DIAGNÓSTICO
A SGB deve ser suspeitada em todos os pacientes com fraqueza motora ou déficit sensitivo não explicado afetando os membros. Os achados típicos no exame neurológico incluem fraqueza progressiva e ascendente e arreflexia tendinosa. É fundamental a pesquisa de infecções prévias. É imperativa a coleta do líquido cefalorraquidiano, que frequentemente mostra dissociação albuminocitológica, ou seja, contagem celular normal com hiperproteinorraquia. Nos casos de pacientes com síndrome de imunodeficiência consequente ao HIV pode haver celularidade liquórica em torno de 10 a 50 células/ml.
Critérios para diagnóstico de SGB
1 – Achados necessários ao diagnóstico
Diminuição de força progressiva em mais de um membro.
Arreflexia tendinosa.
2 – Achados que reforçam o diagnóstico
a) Achados Clínicos
Progressão.
Simetria Relativa.
Sinais e Sintomas discretos.
Comprometimento de nervos cranianos: paralisia facial ocorre em 50% dos casos e frequentemente são bilaterais. Pode acometer nervos responsáveis pela deglutição.
Recuperação: começa geralmente de 2 a 4 semanas após parada da progressão.
Disfunção Autonômica: taquicardia, hipotensão ortostática, sinais vasomotores.
Ausência de febre no início dos sintomas.
b) Achados no líquido cefalorraquiano que dão forte apoio ao diagnóstico:
Hiperproteinorraquia.
Celularidade liquórica normal ou alta no máximo de 10 células mononucleares.
c) Achados que causam dúvidas quanto ao diagnóstico:
Assimetria persistente e marcante da fraqueza.
Disfunção vesical ou intestinal persistente.
Mais de 50 células mononucleares no líquor.
Presença de leucócitos polimorfonucleares no LCR.
Nível sensitivo bem definido.
d) Achados que eliminam o diagnóstico:
História de abusos de hexacarbonos.
Metabolismo anormal das porfirias.
Diagnóstico recente de difteria.
Intoxicação pelo chumbo com neuropatia.
Diagnóstico definido de poliomielite, botulismo, paralisia ou neuropatia tóxica.
SUBTIPOS CLÍNICOS DA SGB
Após o advento da eletroneuromiografia foi possível reconhecer e classificar 4 subtipos da SGB.É notório que a variante Miller-Fischer é um subtipo bem caracterizado clinicamente.
Polineuropatia desmielinizante inflamatória aguda (PIDA)
É a mais comum forma da SGB tendo uma incidência de 85 a 90% dos casos.Esta é considerada uma desordem autoimune desencadeada por uma infecção prévia, geralmente por citomegalovirus e Epstein-Barr vírus. Na PIDA frequentemente há comprometimento dos nervos cranianos, nervos sensitivos e há comprometimento autonômico. A recuperação é geralmente boa e a eletroneuromiografia mostra diminuição da velocidade de condução diminuída em 2 ou mais nervos motores.
Neuropatia axonal motora aguda (NAMA)
É outra forma da SGB, com incidência maior no Japão e China, geralmente devido infecção prévia pelo Campylobacter jejuni. O quadro clínico é semelhante a PIDA, porém raramente há comprometimento de nervos cranianos, nervos sensitivos, e raro comprometimento autonômico. A recuperação pode ser lenta ou rápida. A Eletroneuromiografia mostra degeneração axonal motora pura, com redução da amplitude dos potenciais de ação do nervo, sem desmielinização.A NAMA é frequentemente associada a anticorpo anti-gangliosídeos (GM1, GM1b, GD1, Ga1NAc-GD1a).
Neuropatia axonal sensitivo-motora aguda (NASMA)
Tem baixa incidência, em torno de 10%, com comprometimento motor e sensitivo, detectado na eletroneuromiografia.
Síndrome de Miller-Fischer
É uma variante da SGB, caracterizada pela tríade arreflexia, ataxia e oftalmoplegia. Esta desordem é muita associada a anticorpos anti-gangliosídeo GQ1b.O diagnóstico dessa variante é eminentemente clínico e tem bom prognóstico.
TRATAMENTO
A pessoa acometida pela síndrome deve ser internada em UTI devido ao risco de arritmias e insuficiência respiratória. O tratamento deve ser tanto de suporte como também específico.
TRATAMENTO DE SUPORTE
As principais complicações da SGB são a insuficiência respiratória e hipotensão. Cerca de 25 a 33% dos pacientes evoluem com insuficiência respiratória devida à falência diafragmática e necessitam de entubação orotraqueal e ventilação mecânica; porém este número pode diminuir com a instituição de medicação específica correta.A capacidade vital é o melhor parâmetro para indicação da entubação orotraqueal, quando esta estiver menor de 15mL/kg.
Outra complicação é a hipotensão postural que acomete 10% dos pacientes com SGB, devido à disautonomia, sendo tratado com infusão intravenosa de solução cristalóide e por agentes vasopressores.
TRATAMENTO ESPECÍFICO
Sabe-se, que a plasmaférese e a imunoglobulina (imoglobulina IV) são os tratamentos de escolha para SGB. No entanto, é imperativa a prescrição do tratamento até 2 semanas após o seu início, pois após não há efeito algum.
A droga de primeira escolha é a imunoglobulina 0,4g/kg por 5 dias, pois é menos invasiva que a plasmaférese e tem menos efeitos colaterais. A plasmaférese é feita com troca de plasma na fração de 50ml/kg de peso em 5 sessões separadas por 7-14 dias. A combinação da imunoglobulina e pasmaférese não trouxe vantagem significante.
O corticosteróide seja oral ou em pulsoterapia não é indicado, porém a associação com imunoglobulina pode ser promissor.
O futuro do tratamento está voltado para o beta-interferon, entretanto não há resultados suficientes que justifique o seu uso.
PROGNÓSTICO
O espectro clínico da síndrome de Guillain-Barré é muito amplo, isto é, varia desde a morte até a recuperação total. A melhora clínica, eletrofisiológica e funcional acontece, geralmente, até 18 meses após o início da doença. A maioria das pessoas acometidas se recuperam em três meses após iniciados os sintomas. Portanto, as paralisias provocadas pela doença são reversíveis.
A necessidade de ventilação mecânica e a ausência de melhora funcional três semanas após a doença ter atingido o pico máximo são sinais de evolução mais grave.
Fonte: http://dicionariodesindromes.blogspot.com.br/2009/09/sindrome-de-guillain-barre.html
Alternativa assinalada no gabarito da banca organizadora: D
Alternativa que indico após analisar: D
Nenhum comentário:
Postar um comentário